7 Ücretsiz WordPress Sayfa Düzeni Eklentisi
En İyi Sayfa Düzenleyici WordPress Eklentisi Kullanıcı için bir sayfa tasarlamak çok önemlidir. En iyi WordPress eklentilerini kullanarak sayfalarınızı düzenleyebilirsiniz.…
En İyi Sayfa Düzenleyici WordPress Eklentisi Kullanıcı için bir sayfa tasarlamak çok önemlidir. En iyi WordPress eklentilerini kullanarak sayfalarınızı düzenleyebilirsiniz.…
Başarılı Bir SEO Raporu Nasıl Oluşturulur? Bir müşteri için herhangi bir SEO çalışmasına başlamadan önce, seo uzmanları bir seo denetimi…
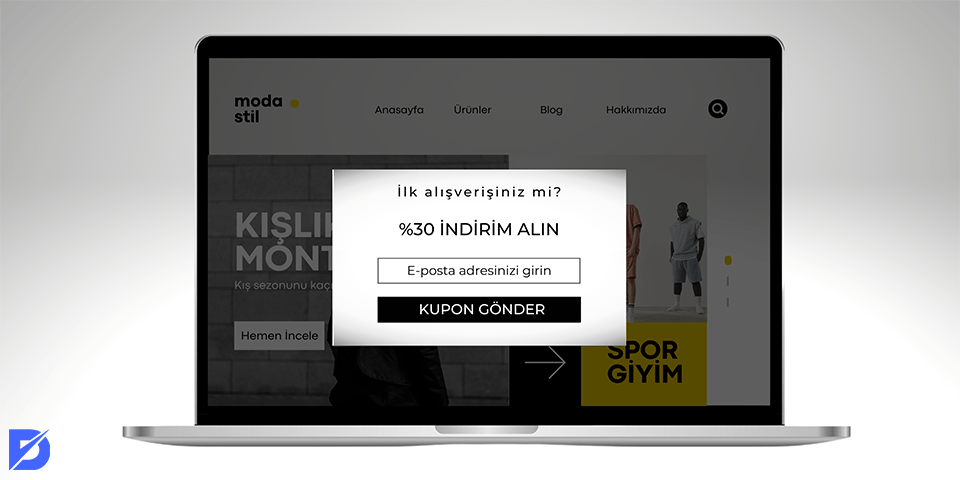
Pop Up Reklamcılık Nedir? Pop up reklamlar nasıl engellenir? Bir web sitesinin sol veya sağ tarafında küçük reklamlar göründüğünde rahatsız…
Sayfa içi arama motoru optimizasyonu nedir? Sayfa İçi Arama Motoru Optimizasyonu Nedir? Sayfa içi ve site içi arama motoru optimizasyonu…
Böcek nedir? Böcek nedir? Oyun oynarken veya internette gezinirken böceklerle karşılaşabiliriz. Böcekler hatalardır. Ancak hatalar yazılımlarda ve diğer programlarda da…
WordPress Güvenlik Eklentileri kılavuzu Web siteniz için güvenlik önlemleri uygulamak üzere WordPress’i kullanın. WordPress güvenlik eklentisi. Bu makale wordpress güvenlik…
Hreflang nedir? Hreflang nasıl kullanılır? Bir Web Sitesine Hreflang Nasıl Eklenir Web siteleri, Google’ı bir web sitesinin dili ve konumu…
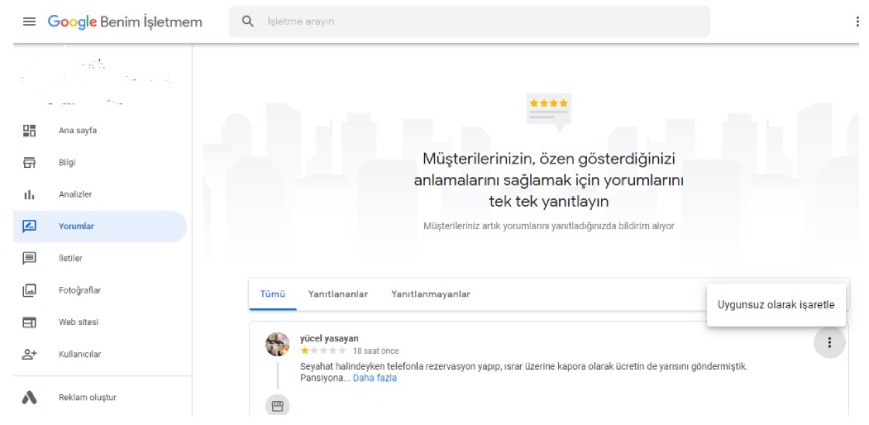
Google’da yorumlar nasıl kolayca silinir? Google Haritalar, Google Hesapları ve İşletmeler için Google Hesapları kullanılabilir. Kötü Google değerlendirmeleri işletme hesaplarından…
Organik Hit Programı Nedir? Organik Hit Programı Nedir? Google’da bunları aradığınızda birçok organik hit bulabilirsiniz. Daha iyi bir alternatif arayınSonuçlar…
Dış SEO nedir? Dış SEO nedir? Site dışı SEO, diğer sitelerden gelen ve backlink olarak da bilinen bağlantıların toplanmasını ifade…